Technologien
Diese Seite ist leider noch nicht auf Deutsch verfügbar.
Key Technologies
Custom Developments
Biochemistry & Proteomics

Functional proteomics is a central component of our research strategy to unravel and characterize components and mechanisms of fast signal transduction processes at membranes which are encoded by constitutive as well as transient or dynamic (membrane) protein interactions forming complexes or extended protein networks.
We have adapted and further developed three major biochemical approaches adressing different aspects of proteins interactions and their dynamics that start from native tissue/cells and use high-resolution mass spectrometry as an unbiased, comprehensive and quantitative readout:
- Affinity-purification mass spectrometry using combinations of antibodies targeting different epitopes (meAP-MS) together with target-unrelated antibodies and target-knockout source material as negative controls to obtain comprehensive and highly reliable interactomes.
- Proximity proteomics (BioID / turboID) to investigate highly dynamic protein-protein interactions.
- BN-PAGE separations for analysis of protein complexes (as 2D-BN/SDS-PAGE or for high-resolution complexome profiling).
- Organellar proteomics (multidimensional subcellular fractionation combined with quantitative MS) to resolve the subcellular distribution of proteins.

Mass Spectrometry
Mass spectrometry allows for highly sensitive and comprehensive identification and quantification of proteins.
We use nano liquid-chromatography-coupled hybrid mass spectrometers (Orbitrap and TIMS-TOF systems) for bottom-up analysis of peptide digests. Acquisition methods are set up to ensure optimal label-free quantification over a broad dynamic range.

Electrophysiology
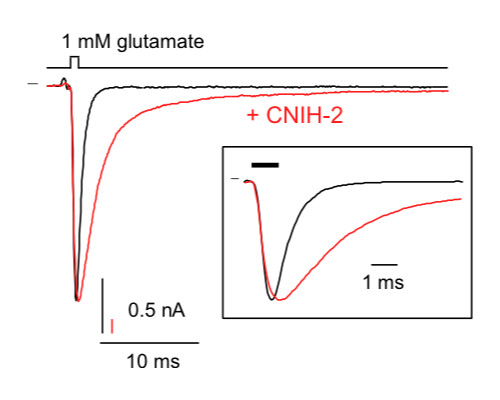
The Patch-clamp technique in its various configurations – whole-cell, cell-attached and cell-free (excised) configuration patches – enables monitoring of function(s) and functional properties of membrane proteins and protein assemblies/complexes with high sensitivity and exquisite resolution in time. In combination with defined genetic manipulations (knock-out, knock-down and site-directed mutagenesis by viruses) the respective recordings promote immediate correlation between the structure and the function of proteins and protein complexes.
We use multiple techniques and recording configurations on neurons in brain slices and neuronal cultures and heterologous expression systems (cultured cells (HEK293, tsA, CHO, COS) and Xenopus oocytes):
- Classical patch-clamp in whole-cell and excised-patch configuration (inside-out, outside-out)
- Giant patch-clamp recording (patch-pipettes with Ø of up to 20 µm)
- Two-electrode voltage- and current-clamp
- Fast piezo-driven application of agonists with sub-millisecond precision
- Whole-cell recordings combined with fluorescence microscopy in confocal and 2-photon (2 pi) configuration (for excitation of individual synapses or axons)

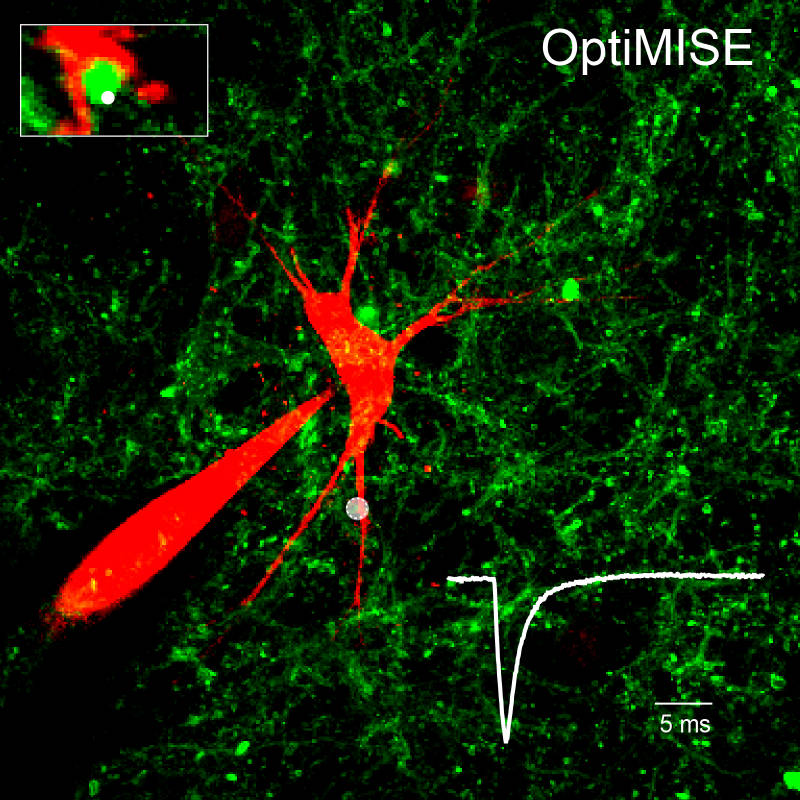
by action potentials triggered in paired (pipette 1) and 2pi (channel rhodopsin) configuration.
Cell Biology & Fluorescence Microscopy
Accurate assessment of the expression profile of proteins and protein complexes, as well as of the morphology of cells and sub-cellular compartments are fundamental for understanding cell biology and dynamics of any type of cell.
We apply high-resolution confocal fluorescence-microscopy in various configurations in combination with antibody-based immuno-cytochemistry, dye-infusion into defined cells and 3D-reconstruction from individual images to determine/characterize
- Expression and localization of target proteins across the entire brain, in defined brain regions and specific types of cells and subcellular compartments
- Dynamics of protein expression and distribution following genetic manipulations (constitutive and virally-driven) and during postnatal development
- Morphology (shape and organization) of cells and sub-cellular compartments (including synapses) and their dynamics



Electron Microscopy
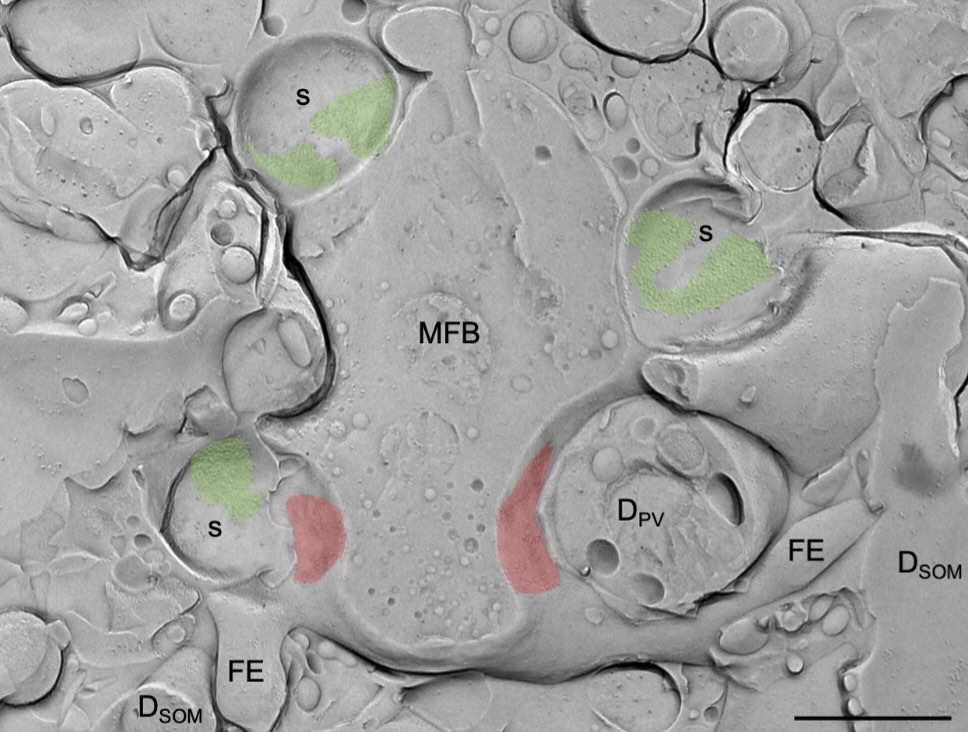
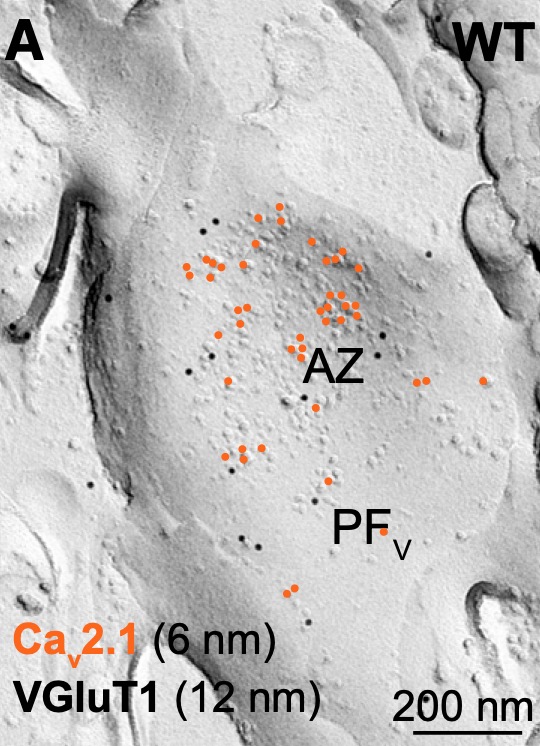
Rapid and reliable information processing in neuronal circuits depends on the compartmentalization of and functional interaction of signaling proteins, as well as the activity- and input-dependent changes in molecular and morphological properties of central neurons. Using high-resolution conventional and quantitative SDS-digested freeze-fracture replica labeling immunoelectron microscopy, as well as Focused Ion Beam Scanning Electron Microscope (FIB/SEM) and SEM Array Tomography we study the distribution and dynamics of surface abundance of pre- and postsynaptic neurotransmitter receptors, ion channels and other functional molecules, as well alterations in dimensional parameters and synaptic architecture of cortical and subcortical neurons.

Bioinformatics
Modern scientific research heavily depends on information technology to support and accelerate the process of converting experimental data into knowledge.
Our efforts are focused on advancing proteomics research, specifically analysis of large amounts of mass spectrometry (MS) data with the aim of identifying and quantifying proteins and their interaction partners. For this, we
- develop customized algorithms and software for (a) solving complex problems, (b) optimal processing of experimental data and (c) visualizing the results
- develop and operate fast data analysis pipelines
- run our own servers and service infrastructure (physical, virtual, containers)
- evaluate, integrate and operate third-party software.
Custom Developments
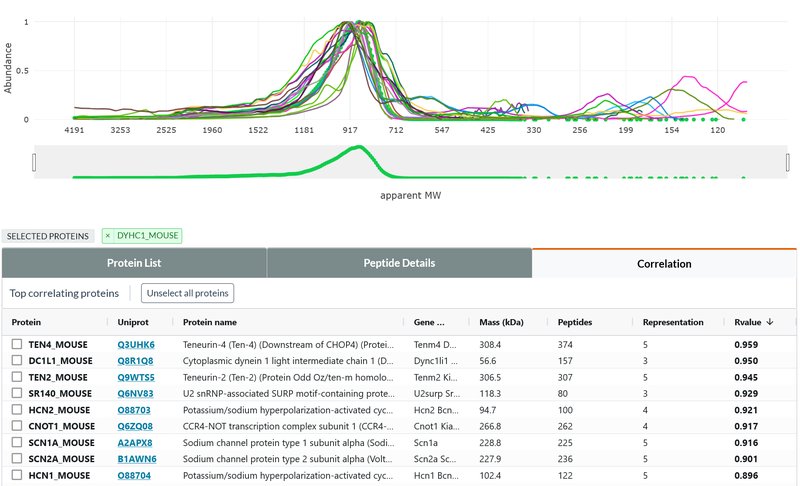
Complexome Profiling
As an untargeted, large-scale alternative approach, we have developed a unique high-resolution complexome profiling technique that is based on (i) separation of gently solubilized proteins (complexes) on a native polyacrylamide gradient gel, followed by (ii) sub-mm sampling with a cryo-microtome and (iii) quantitative mass spectrometric analysis (csBN-MS). The highly resolved abundance / size distribution profiles obtained for thousands of proteins reflect their complex molecular organization on a systemic level. Interpretation of these rich datasets will be fostered by future development of powerful bioinformatic tools. Identified protein entities are further investigated/corroborated by meAP-MS analysis or structural methods (like SPA).

Multi-Epitope Affinity Purification-Mass Spectrometry (meAP-MS)
For targeted analysis of protein-protein interactions we use target-specific antibodies for solubilized target (complex) purification and subsequent analysis by LC-MS/MS mass spectrometry. Importantly, to overcome adverse reactions (like cross reactivities, selection biases or disruption of target assemblies) of individual antibodies, we use combinations of antibodies targeting different epitopes (meAP-MS) together with target-unrelated antibodies and target-knockout source material as negative controls. Upon quantitative MS analysis with broad dynamic range (typically more than 3 orders of magnitude), co-purification specificity, efficiency and consistency of proteins are stringently evaluated using our Belki software tool to obtain comprehensive and highly reliable interactomes.

Membrane protein solubilization
Biochemical analysis of membrane proteins represents a particular challenge since they require the use of detergents that inherently compromise between high solubilization efficiency and preservation of structural integrity of proteins and protein assemblies. We systematically investigate solubilization conditions in different biochemical assays (Western blot analysis, native gel electrophoresis, binding assays) benefitting from the longterm know-how and standardized ComplexioLyte buffers from our collaboration partner Logopharm.
Organellar proteomics
To investigate the subcellular distribution and dynamics of proteins at systemic level we adopted a protein co-fractionation approach commonly referred to as organellar proteomics. Specifically, we subject cells or tissues to multi-dimensional fractionation e.g. by stepwise homogenization / lysis, density gradient and sedimentation ultracentrifugation typically resulting in 40-100 samples that are analyzed by quantitative mass spectrometry. The preferred subcellular localisation of proteins can then be investigated by clustering of their abundance distribution profiles with established subcellular markers (for example using our Belki software).
In addition to these general approaches, we use a variety of specialized techniques for biochemical characterization of selected proteins/protein complexes:
- Two-dimensional native/SDS-PAGE (including antibody shift assays)
- Analytical protein chromatography (e.g. size exclusion or affinity chromatography)
- Reconstitution of complexes by co-expression in heterologous systems
- Multi-step purification of reconstituted protein complexes for single particle analysis (cryo-EM, in collaboration)
- Analytical ultracentrifugation
- Advanced sample collection techniques (e.g. tissue micro-puncher, microscopy-assisted laser-dissection, FACS sorting).
- Chemical labelling / cross-linking of proteins